Yates Laboratory at The Scripps Research Institute is Pushing for Better Understanding of Cystic Fibrosis with the help of Cloud HPC on AWS with CloudyCluster and IP2
On Oct 10, 2018
Cystic Fibrosis (CF) is one of the most common inherited childhood diseases, impacting 1:4,000 children born in the US (www.cff.org). CF is caused by mutations in the gene that encodes the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein, which is responsible for regulating the flow of salt and fluids in and out of the cells in different parts of the body. Mutations to the CFTR gene produces a defective protein that is unable to fold into the conformation necessary for proper function.
Through funding from the NIH (Grant ID: 1 R01 HL 131697-01A1), the Yates Laboratory at The Scripps Research Institute in La Jolla, led by professor John Yates, is actively pursuing a better understanding of this fatal disease. To aid in their efforts, the Yates Laboratory has taken advantage of the expansive computing capacity of AWS and CloudyCluster to provide High Performance Computing (HPC) that can speed up their computational investigations. By using a traditional computing cluster, this project required over a hundred thousand core hours, but CloudyCluster was able to simplify the computational process and significantly reduce the investigation time by moving the workload to the cloud.
Background
To better understand this project, let’s start with a little background. Studies on the genetic code started after the structure of DNA was proposed in 1953. The determination that the information in DNA was transcribed into RNA and subsequently translated into protein was one of the great achievements of the last century. The finding that the genetic code was universal in all organisms was surprising and significant. Scientist had discovered the Rosetta Stone to biological information (aka “cracking the genetic code”). But, as with the Rosetta Stone, there was a lot of work left to decode the details. It was soon apparent that there was actually far more to understand. After the translation of the RNA message into proteins they must fold into the proper conformation and proteins are modified with many different types of Post Translational Modifications (PTM) that are necessary to achieve and modulate biological activity. These PTMS are a protein code that controls cellular functions. A component of protein function is determining how modifications are used to regulate or alter function.
One of the methods used to investigate the function of PTMs is to research genetic mutants, such as those CFTR mutants that cause the human protein misfolding disease Cystic Fibrosis. Investigating PTM processing in the mutant CFTR protein may lead to a way to restore function, and thus potential therapies. It may also provide insight into more general mechanisms of how PTMs contribute to protein misfolding diseases, such as Alzheimer’s Disease or Parkinson’s Disease.
Investigation
The majority of Cystic Fibrosis cases are caused by a deletion of phenylalanine 508 (∆F508) in the CFTR protein. This deletion leads to an altered 3D structure of CFTR, resulting in loss of anion channel function, because the CFTR protein does not reach its proper location in the cell membrane. Furthermore, initial studies by the Yates lab indicated that post-translational modifications of the misfolded ∆F508 CFTR protein are different from wt CFTR. Identifying CFTR PTMs, especially differentially regulated ones, is of enormous importance to better understand the ∆F508 CFTR channel and cellular processing defects and could provide new strategies for therapeutic intervention.
Identifying CFTR PTMs comprehensively is difficult because the concentration of CFTR in cells is very low and many different PTMs and combinations of PTMs are possible per protein. And while some PTMs may be quite abundant, others are just above the detection level. An unbiased search for PTMs present on the protein (“Blind-PTM search”) has the potential to identify all PTMs (including SNPs and amino acid deletions) because peptide sequences are identified along with peptides that deviate from predicted molecular weights, signaling the presence of modifications. However, identifying PTMs by searching extracted tandem mass spectra against the entire human genome (Blind-PTM) is computationally demanding. Such queries can take up to two weeks on 600 node cluster for a single experiment. [1][2] CloudyCluster HPC cluster on AWS enables higher parallelization and can significantly reduce run-times.
Resources
A Blind PTM search is computationally expensive due to the large search space it requires. The Yates Laboratory requested hundreds of thousands of core hours on AWS, leveraging CloudyCluster through the NIH Cloud Credits pilot program. The required software was easily added to the base CloudyCluster AWS Marketplace ami and, when launched, a familiar HPC environment was available with similar functionality to what was available at Scripps. This enabled the AWS computational resources to help resolve the search bottleneck and accelerate the time to results.
Applications/Tools
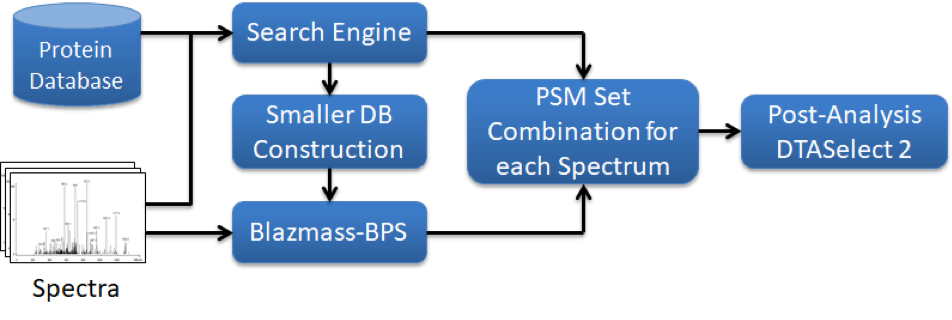
The Yates Laboratory developed the Blind-PTM search engine to identify unknown PTMs and allow for an unbiased search approach for PTMs. To reduce the large search space, spectra that could be identified by traditional forward searches were excluded and only unidentified spectra were included in the Blind-PTM search. However, even with the reduced number of spectra, the search is very expensive to run on traditional computers. For cloud job submission, the Integrated Proteomic Pipeline (IP2) cloud module leveraging AWS and CloudyCluster was used. The IP2 is also currently being used at a variety of research labs, universities, pharmaceutical companies and research hospitals.
Datasets and Results
Over 90 independent samples were analyzed on high mass accuracy mass spectrometers such as the LTQ-Orbitrap Elite (Thermo Fisher). Each experiment generated > 1 GB of data and contained about 15,000 - 20,000 spectra that were identified by the traditional ProLuCID search approach. While the traditional search approach identified around 80,000 CFTR spectra, and revealed important PTMs that were quantitatively and qualitatively altered in Cystic Fibrosis, the Blind-PTM search revealed an additional 30,000 CFTR spectra that contained various PTMs. Ultimately, it was the Blind-PTM search approach that revealed additional critical information on PTM changes that would have been missed by traditional searches. The Yates lab is currently following up on several of these PTM changes to see if influencing some of these PTMs could aid in CF therapy and a paper describing the results will be published soon.
Long-term plan
The Scripps Research Institutes has significant HPC computational resources, but to reduce the strain on the shared computing resources the Yates Laboratory is looking to use AWS with CloudyCluster as an alternative solution for big data analysis. Additionally, the Yates lab is looking toward GPUs to continue to speed up this process, with the latest GPUs available in AWS and CloudyCluster the time to results will continue to accelerate.
Write-up Contributors
Robin Park, Sandra Pankow, Titus Jung, Ken Wilson
Relevant References
- Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science. 1992;256(5058):774-9. PubMed PMID: 1375392.
- Pankow S, Bamberger C, Calzolari D, Martinez-Bartolome S, Lavallee-Adam M, Balch WE, Yates JR, 3rd. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature. 2015;528(7583):510-6. PubMed PMID: 26618866.
- McClure ML, Wen H, Fortenberry J, Hong JS, Sorscher EJ. S-palmitoylation regulates biogenesis of core glycosylated wild-type and F508del CFTR in a post-ER compartment. Biochem J. 2014;459(2):417-25. Epub 2014/01/31. PubMed PMID: 24475974; PubMed Central PMCID: PMC3993085.
- McClure M, DeLucas LJ, Wilson L, Ray M, Rowe SM, Wu X, Dai Q, Hong JS, Sorscher EJ, Kappes JC, Barnes S. Purification of CFTR for mass spectrometry analysis: identification of palmitoylation and other post-translational modifications. Protein Eng Des Sel. 2012;25(1):7-14. Epub 2011/11/29. PubMed PMID: 22119790; PubMed Central PMCID: PMC3276306.
- Townsend RR, Lipniunas PH, Tulk BM, Verkman AS. Identification of protein kinase A phosphorylation sites on NBD1 and R domains of CFTR using electrospray mass spectrometry with selective phosphate ion monitoring. Protein Sci. 1996;5(9):1865-73. Epub 1996/09/01. PubMed PMID: 8880910; PubMed Central PMCID: PMC2143550.
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, Wong CC, Fonslow B, Delahunty C, Gao Y, Shah H, Yates JR, 3rd. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics. 2015. PubMed PMID: 26171723.
- Chick JM, Kolippakkam D, Nusinow DP, Zhai B, Rad R, Huttlin EL, Gygi SP. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol. 2015;33(7):743-9. PubMed PMID: 26076430; PubMed Central PMCID: PMCPMC4515955.
- Washburn MP, Wolters D, Yates JR, 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242-7. PubMed PMID: 11231557.
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, Wong CCL, Fonslow B, Delahunty C, Gao Y, Shah H, ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity, J Proteomics. 2015
Cover Image Credit: https://commons.wikimedia.org/wiki/User:Emw under CC BY-SA 3.0